
 Yahoo奇摩新聞
Yahoo奇摩新聞 罕病律師陳俊翰40歲驟逝!健保最貴藥物1劑4900萬就用在此病
罹患罕見疾病脊髓性肌肉萎縮症的律師陳俊翰,春節期間因感冒引發併發症病逝,得年40。兩年前陳俊翰放棄美國對罕病病患周全的照護,返台與為病友爭權益,如今英年驟逝令人不捨。
什麼是脊髓性肌肉萎縮症?
高雄醫學大學附設中和紀念醫院小兒部暨檢驗醫學部教授鐘育志表示,脊髓性肌肉萎縮症(SMA,全名為Spinal Muscular Atrophy)是因SMN1基因缺失或突變,導致運動神經元退化,造成肌肉無力及萎縮,「這是一種隱性遺傳性疾病,」即使家族有此基因,也不一定每個人都會發病。
SMA不是漸凍症,是嬰幼兒死亡率最高的罕病
過去社會大眾缺乏對於SMA的正確認識,很多人以為這是漸凍症,鍾育志表示,SMA與「漸凍症」(Amyotrophic Lateral Sclerosis,簡稱ALS)容易混淆,兩者雖然都跟運動神經元退化有關,但漸凍症好發年齡介於50至75歲,SMA病友在18歲之內發病者高達99%。
據統計,台灣目前大約有420名SMA病友,大人小孩皆有;平均帶因率約2.1%,估計台灣有將近50萬人為無症狀的SMA基因者。
「一旦確診SMA,主要臨床表現包括肌肉張力低下、四肢與軀幹肌肉無力及肌肉萎縮,而且運動功能會隨著年齡增長持續退化。」鐘育志說,坐輪椅的病友幾乎都是極重度肢體障礙,病情嚴重者,除了肌肉沒有力量,還有一個特徵,就是全身肌肉鬆軟到頭部垂下都無力撐起,不但照顧者難以抱起,病友可能連自己移位、如廁、刷牙、洗臉、拿碗、舉筷、咀嚼、吞嚥都有困難,甚至睡覺翻身或呼吸的力氣也沒有,晚上還需使用呼吸器。
SMA依發病年齡與最佳運動功能將疾病分為4種類型,其中約6成SMA病友在出生6個月以內發病,是最嚴重的第1型,若未及早治療,2歲前死亡率高達8成,是全世界嬰兒死亡率最高的罕見遺傳疾病。

陳俊翰是最嚴重的第1型病友,卻做到出國攻讀碩博士
陳俊翰就是SMA第1型病友,出生後就全身無力,到1歲還無法抬頭、翻身、獨坐,後來就醫檢查確診罹患SMA,當時無藥可醫,醫師評估他僅能活3~4年。他免疫力差,小時候三天一小病、五天一大病,大半時間在醫院及家裡,也因為運動功能退化,每次用餐往往超過1個半小時。
陳俊翰國小六年級時病情惡化,在加護病房插管長達2個月,一度失去求生意志,寫紙條給媽媽說「不要救我,我真的撐不下去了。」在媽媽陪伴下,他跨過心中關卡,轉念思考「家人都不輕易言棄,自己更不應該放棄。」
陳俊翰媽媽最初曾怨懟命運對她及孩子不公,但為了孩子她很快打起精神、不辭勞苦帶陳俊翰四處求醫,甚至遠赴日本、美國找尋解方。40年來,陳媽媽不但是陳俊翰的手跟腳,協助日常起居,更是兒子實踐夢想的最佳助手。
隨著肌肉力量被剝奪,陳俊翰幾乎24小時都需要有人在旁協助,「大部分是媽媽在旁邊幫助我。」陳媽媽一直告訴陳俊翰:「你行動不方便,我做你的手腳,你沒辦法的部分我會幫你補足,你盡自己的能力,能夠做多少算多少。」
然而陳俊翰做到的,遠遠超過旁人的預期。他國中時病況惡化到無法坐立,做不到翻書閱讀,只能躺在床上聽錄音帶自學。行動不便沒有禁錮他的心靈,他每天讀書的時間比同學少,「那我就用加倍的努力來彌補!」從小他用3倍時間、3倍努力保持優異成績,大學考上台大,雙主修會計和法律,11年前他遠赴美國哈佛攻讀法學碩士、在密西根大學取得法學博士學位,並在柏克萊大學做了1年訪問研究。
放棄留在美國,返台為病友爭權益
美國的保險制度對於罕病病友照護完善,SMA用藥全額給付,陳俊翰原本可繼續留在美國獲得完善治療與照顧,但看見國內有許多罕病用藥尚未給付,加上親身體會美國願意給罕病病友最基本的尊重、珍惜每一條生命的價值,他選擇回台,希望運用自己的力量推動罕病用藥權益。
「哪怕讓多一點人有機會接受用藥,也會比我自己一個人在國外獲得治療更有價值。」陳俊翰抱持這樣的信念,2022年放棄美國高薪工作返台,除了是中央研究院法律學研究所博士後研究學者,身為台灣首位SMA病友律師,他同時擔任中華民國肌萎縮症病友協會及社會法人台灣弱勢病患權益促進會的法律顧問,致力罕病與身障病友權益的推動。
從無藥可醫到有藥可選,全球最貴罕病藥去年納健保
早年SMA是沒有治療藥物的神經退化性疾病,病友家庭也因為不了解疾病進程及醫療資源在哪裡,在照護上面臨種種考驗。1987年,鐘育志以肌肉切片輔助診斷出台灣第1個SMA案例,自此積極投入SMA轉譯研究與整合照顧,迄今37年,歷經從「無藥可醫」到「多藥可選」,見證醫學的進步,他因此被稱為「台灣SMA之父」。
「2016年開始,SMA才陸續出現治療藥物,」台大醫院神經部教授蔡力凱表示,在無藥可醫的年代,臨床主要依據病人的需求提供營養與呼吸照護,預防肌肉無力產生的併發症。後來治療藥物開始研發,台灣醫療團隊積極參與全球SMA藥物臨床試驗;藥物問世後,台灣成為全球第1個全面實施SMA新生兒篩檢且提供確診嬰幼兒藥物治療的國家,更陸續擴大健保給付的藥物種類與對象。
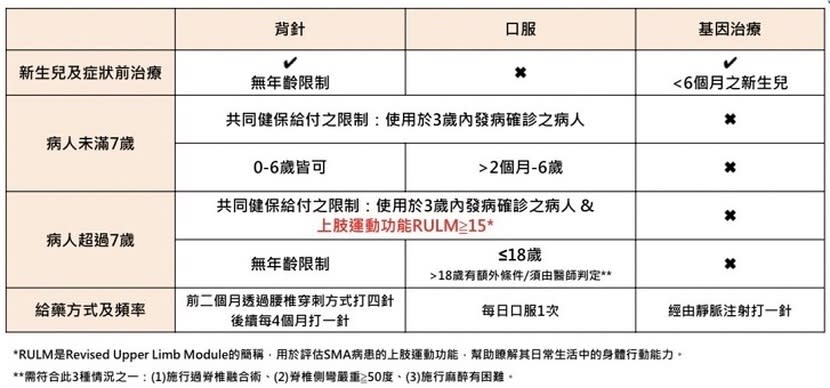
目前SMA治療藥物包括針劑(因施打部位為背部腰椎脊髓腔,俗稱「背針」)、口服藥與基因治療等3種新興治療藥物,透過影響特定基因表現,不但有助減緩惡化、阻止退化,「幫病程踩煞車」,讓病患免於運動功能退化的恐懼,同時持續穩定呼吸進食功能,臨床上更有病友因及早治療,運動功能出現進步。
比如鐘育志的小病人、9歲的晴晴是第1型SMA,從小常因小感冒引發嚴重肺炎,數次呼吸衰竭險些致命,為了保命,晴晴每天24小時配戴呼吸器,足不出戶,4歲半時接受藥物治療後,依賴呼吸器的時間漸漸縮短,現在只有晚上睡覺時需配戴呼吸器,白天可以跟家人出門旅遊、露營。
台灣脊髓肌肉萎縮症病友協會現任理事長黃旭輝是第3型SMA病友,苦等50多年後,去年開始接受治療,開心發現失去的體力回來了,晚上睡覺可以獨力翻身,不用一再吵醒太太幫忙翻身。
健保自去年(2023)8月起將SMA基因治療藥物「諾健生(Zolgensma)」納保,病友終身只需注射1劑,但1劑高達4,900萬元,不但是健保目前最貴的藥物,也是全球最貴的藥物之一。只有「6個月以下發病,且帶有基因突變」的小病患可用,預估一年有8名病童受惠。南部一個未滿6個月大的嬰兒,於去年8月8日,成為全台第1個使用此藥物的患者。
健保給付3種SMA治療藥物
SMA依照發病年齡和可達到的最佳運動功能分型

病友跟時間賽跑,有藥卻無法治療比沒藥更殘忍
「SMA愈早治療效果愈好,如果能在發病前開始治療,病人的運動能功能發展幾乎可保持正常,」蔡力凱說,目前3種治療藥物健保有條件給付,但有約300名SMA病友,或因上肢運動功能評分未達給付標準,或因疾病惡化速度較慢,被排除在健保給付之外。根據文獻,即使目前病情較嚴重,用藥也有機會改變疾病進程,疾病進程較慢的病患及早治療,才能維持現有運動功能。
天價的基因治療打1針即可,背針及口服藥則必須一輩子用藥,費用也高昂。蔡力凱指出,背針打1針大約250萬元,第1年要打6針,之後每年打3針;口服藥必須每天服用,1年藥費700多萬元。非一般家庭能負擔,希望政府給病友機會。
農曆春節前陳俊翰出席「SMA脊髓性肌肉萎縮症媒體工作坊」時表示,每一位病友都在跟時間賽跑,迫切需要藥物阻止病情惡化,「人權的意義就是生而為人應該享有的權利,」一個人身體是不是還能夠活動、手是不是還能舉高,都不影響到他的尊嚴跟價值。對這300名病友來說,有藥卻無法治療,比沒藥更殘忍。
「期待每個罕病病友都享有平等醫療權益」是陳俊翰放棄小我,返台推動罕病病友用藥權益的初衷,他雖然提早抵達人生終點,但他的故事和精神鼓舞了罕病病友,繼續在生命之路上奮鬥。
延伸閱讀:
